




 免费会员
免费会员 进入店铺
进入店铺 店铺留言
店铺留言




1.依照人體細胞組織優良操作規範(Good Tissue Practice, GTP)打造

國璽幹細胞公司致力於推動台灣再生醫學,並在生醫園區裡依照人體細胞組織優良規範(Good Tissue Practice, GTP)打造超高規格潔淨實驗室,製作出符合人體臨床試驗所需要的各種規格幹細胞製劑。
幹細胞製劑中心所有軟硬體皆遵守GTP規範的要求,GTP規範是為了預防因使用人體組織細胞進行研究過程中傳播或擴散傳染病,協助確認所使用的人體細胞組織物不含任何傳染病源且在製造過程中不受污染。
2.具備「Class 10,000等級」超高規格無塵環境
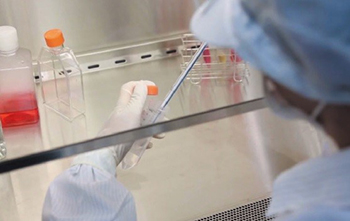
無塵等級比一般手術室的要求還高,以超高規格符合國際標準。研製中心內採用專業用燈具,確實做到不沾灰、無落塵,而地板完全無接縫,牆面與地板交接處為圓弧形,不易藏污納垢,避免污染。另外處理幹細胞製劑的潔淨實驗室是由各系統組成,包含環氧樹酯塗裝、高架地板、隔間金屬庫板、天花金屬板、高效空氣微粒過濾器(HEPA)、乾濕冷盤、水風管、公用系統管路、循環空調箱、外氣空調箱等元件,才可打造出氣密的高標準,生產出高品質產品。
3.採用MAU(Make-Up Air Unit)空調系統維持「正壓」、「恆濕」、「恆溫」

研製中心使用的空調系統構型為抽風式外氣空調箱(MAU),在無塵室內為了維持室內正壓、外氣微塵粒過濾及潔淨室內相對溼度的控制以及避免受到外界環境污染,因此必須透過MAU空調系統隨時補充足夠外界新鮮空氣。此外,無塵室內人的呼吸、污染物質濃度的稀釋、濕度控制…等都依賴外氣來對無塵室作微調功能,因此無塵室MAU空調系統對無塵室之功能像人類呼吸一樣重要,每個高精密產業都必須要有很好的MAU(Make-Up Air Unit)空調系統。
4.以淨、污分離通道降低實驗室與外界污染的可能

國璽幹細胞公司將實驗人員出入通道嚴格劃分,實驗時人員需著換無塵衣、無塵鞋並徹底殺菌後再由淨走道入實驗室工作,實驗完畢後必須從汙走道離開實驗室,且無法逆行回實驗室,以確保單向進出實驗室,降低不必要的汙染。
5.配合醫療級HEPA高效空氣微粒過濾器,有效減少空氣中外來微粒及微生物
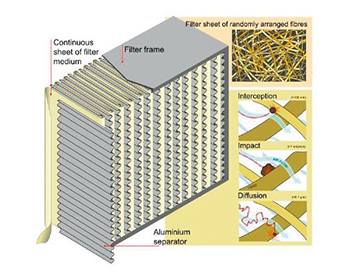
為確保空氣中微塵粒子數量處於絕對穩定的狀態下,經由空調系統使得進入製劑中心內的空氣再經由醫療級HEPA (High Efficiency Particulate Air) 高效空氣微粒過濾器過濾後,能有效預防細胞處理過程中受到外來汙染,HEPA對於0.1微米和0.3微米的微粒及微生物的有效過濾率高達99.998%。
6.具Class 100等級的高級進口生物安全櫃,可保護細胞、操作者及環境安全。

國璽幹細胞公司使用比一般無菌操作台更高級的生物安全櫃進行實驗,它具有特殊層流風向設計及HEPA高效微粒空氣過濾,藉由櫃體內的高效濾網過濾進排氣並在櫃體內產生向下氣流的方式避免交叉汙染,確保細胞及人員安全。研製中心每次實驗前會先以酒精進行消毒清潔,而實驗後會以UV紫外線滅菌燈照射進行滅菌、有效破壞微生物DNA與RNA結構。
7.採用傳遞箱讓人、物分道

所有要進入無塵室的檢體、耗材以及物料傳遞都需經由傳遞箱(Passing Box)送入無塵室,避免人員進出頻繁而破壞無塵室內壓力,引起微粒進入。
8.採用原裝進口全自動過氧化氫燻蒸滅菌型二氧化碳培養箱

擁有創新的內建過氧化氫燻蒸滅菌裝置,滅菌時間短且不殘留有害化學物質,能有效殺滅微生物防止交叉汙染。不採傳統高溫滅菌法,提高設備穩定性及可靠度,保證控制係統靈敏性和腔內潔凈度,確保細胞培養安全。全不銹鋼隔板,腔內全圓滑轉角設計,便於清潔。
9.採用臨床應用、安全性高的抗污染隔氮型儲存設備

幹細胞儲存是採用隔氮式儲存設備,可持續維持在-190℃以下,並確保樣品不會與液態氮直接接觸,避免細胞遭受汙染。全智慧自動調控儲存設備內液態氮含量,確保槽內溫度恆定於極低溫,穩定幹細胞的保存狀態。
10.高標準無菌檢驗程序與設備,以確保幹細胞處於無菌狀態

為確保幹細胞生產過程中的感染控制,依照藥典方法檢測如:無菌試驗、黴漿菌檢測、內毒素檢測等。並制定相關的檢測程序,相關試劑與設備定期查驗及校正,以確保幹細胞無汙染。
11.投保火災綜合保險,提供最完善的保障
為確保實驗樣品因意外而受到影響,國璽幹細胞公司針對實驗室投保火災綜合保險,提供完善的保護。
12.24小時全天候作業環境監控系統,若遇異常可即時通報並解決
全天候作業環境監控系統可隨時掌控儲存設備的最新狀態,在每一個時間透過電腦監控所有位置的溫度、濕度和壓力,保持恆定狀態,一旦發生異常狀況,會在第一時間自動發出警示並即時通知專責人員處理。
幹細胞生長因子產品開發中心-創造人類再生夢想的無限可能
1.依照實驗室優良操作規範(Good Laboratory Practice, GLP)打造分子生物實驗室

實驗室優良操作規範 (Good laboratory practice, GLP) 係指有關實驗室試驗之計畫、執行、監測、記錄、報告及檔案的組織架構及規範。目的在於確保實驗結果的可追溯性,確保實驗結論的品質及可信。為了研究幹細胞在分子層面上的生理行為上的,產品開發實驗室擁有各種分子生物及細胞生理研究所需之儀器設備,其中包括細胞培養室、無菌操作台、螢光顯微鏡、超高速離心機、電泳設備、影像分析系統、分光光度計、免疫分析儀、藥品抽氣櫃等。
2.具備「Class 10,000等級」超高規格細胞培養實驗室

為了改良細胞製程及研發應用技術,細胞培養實驗室的潔淨度規格完全相同於幹細胞製劑中心,細胞培養實驗室可進行幹細胞取得,並長期體外培養以做為研究用途的過程。目前,不論貼附細胞株、懸浮細胞株以及初代細胞均能在本實驗室成功的培養生長。wei xin:wlayar細胞培養實驗室的應用,非常廣泛,諸如:大量細胞培養製程的建立、藥物開發以及許多生物科技產品。本實驗室的設備包括:低溫離心機、無菌操作台、二氧化碳培養箱、倒立螢光顯微鏡、液態氮儲存槽等設備,提供幹細胞培養的技術開發。
3.全自動高溫滅菌二氧化碳培養箱

細胞培養實驗室擁有創新的180℃高溫滅菌,滅菌時間最短。專利銅制隔離室,於高溫滅菌時能自動保護CO2傳感器,無需拆卸紅外傳感器,保證控制係統靈敏性和腔內潔凈度。具有超溫保護裝置,確保細胞培養安全。全不銹鋼隔板,腔內全圓滑轉角設計,便於清潔。
4.高淨化純水系統

純水系統可從自來水連續生產純淨的二次水,符合CAP、NCCLS和ISO3696/BS 39二次水標準,純化過程的每一個步驟中都內置了對水質純化進行控制的技術,保證了使用時水質的穩定。
5.雙雷射四螢光式流式細胞儀

雙雷射四螢光式流式細胞儀配備有一支藍光雷射和一支紅光雷射、兩個散射光檢測器,並載入了預先最佳化光學濾光片的四個螢光偵測器,緊湊的光路佈局、固定式的光路設計和經過預先最佳化的檢測器設置使整套系統更加易於使用,且資料數位採集具有跨越7個數量級的動態範圍 (1600萬道的數值化資料解析度),能保證所有資料總是有效可用的。
6.研究級倒立螢光顯微鏡

研究級倒立螢光顯微鏡上使用了全新的UIS2光學系統。這種新型的光學系統有著極高的信噪比,在更寬的波長範圍內消除色差,有著平坦的高透射率。該系統建立了全新的螢光性能標準,不必損傷細胞,就能有效地檢測到微弱的螢光信號,並且強化了
YAO物非临床安全性评价产生的背景:
国际经济合作与发展组织(OECD)
提出了新化学物上市前的最低限度的安全性评价项目
制定了一系列的毒性实验准则
凡按此实验准则进行的毒理学实验,成员国间相互承认和接受
第1节 YAO物非临床安全性评价:
YAO物非临床安全性评价
指在人临床实验之前的毒理学实验研究,即按照规定的毒理学程序和方法评价YAO物对机体产生的有害反应,并外推到通常条件下应用YAO物对人体和人群的健康是否安全。
研究内容:评价YAO物的毒性反应
判断剂量和时间与毒性的关系:最小中毒量、最小致死量、毒性反应的起始和持续时间
确定YAO物毒性的靶器官、中毒机制、毒性反应的性质与可逆性、毒性剂量与安全范围
YAO物非临床安全性评价的目的
:
1.提供YAO物安全性的实验数据,支持I期临床研究。
2.确定临床实验的安全性起始剂量,以及可接受的安全剂量范围。
3.通过广泛检测,发现任何潜在和未知的毒性。
4.在YAO物开发中获得最大的利益/风险性比率。
YAO物非临床安全性评价的局限性:
1.人与动物的种属差异,反应的敏感性不同,甚至有质的差异→假阳性、假阴性;如,沙利度胺。
2.毒理学实验给药剂量较大,存在高浓度向低浓度外推的不确定性。
3.实验动物数量有限,对发生率较低的毒性难于发现→小数量动物向大规模人群外推的不确定性。
4.实验动物遗传背景、身体状况一致,而临床用药的人群个体差异较大。
第2节 YAO物安全性评价GLP实验室
:
非临床安全性研究质量管理规范(good laboratory practice for nonclinical laboratory studies, GLP)
GLP法规的颁布
1999年10月,SDA颁布了《YAO物非临床研究质量管理规范(试行)》。
2003年6月, SFDA局长2号令《YAO物非临床研究质量管理规范》
GLP的适用范围
:
各国的GLP适用的范围不尽相同
美国FDA——色素和食品添加剂、饲料添加剂、人用药品、兽用药品、生物制品、人用医疗器具、电子产品等
经济合作与发展组织OECD——化学制品如工业化学品、人用药品、兽用药品、化妆品、食品添加剂及农药等
中国SFDA——药品
实施GLP的目的:
GLP已成为国际上YAO物安全性试验研究共同遵循的规范
世界各国的GLP虽然各有特点,但是基本原则是一致的
提高药品非临床研究的质量
确保试验数据的真实性、完整性和可靠性
最大限度地避免人为因素产生的错误和误差,尽可能在试验早期发现并修正
保证临床用药安全
YAO物GLP认证试验项目:
单次和多次给药毒性试验(啮齿类)
单次和多次给药毒性试验(非啮齿类)
生殖毒性试验(I段、 II段、III段)
遗传毒性试验(Ames、微核、染色体畸变、小鼠淋巴瘤试验)
致癌试验
局部毒性试验
免疫原性试验
安全性药理
依赖性试验
毒代动力学试验
YAO物GLP认证检查主要内容
组织机构和人员
实验设施与管理
仪器设备和实验材料
标准操作规程
研究工作的实施
资料档案
其它(实验技术现场考核、计算机系统)
申请试验项目
GLP实验室申请的资料审查内容:
一.机构概要
:机构发展概况
组织机构框架图
实验设施平面图
二、组织机构的设置与职责
三、机构人员构成情况、人员基本情况以及参加
培训情况
四、机构主要人员情况
机构负责人
质量保证部门负责人
专题负责人
动物饲养管理负责人
组织病理学检查部门负责人
资料保管负责人
供试品管理负责人
其他负责人
五、动物饲养区域及动物试验区域情况
动物设施面积和动物收容能力情况
各动物饲养区的平面图
动物饲养区人流、动物流、物品流、污物流、空气流等流向图
环境条件
饲料、饮水、垫料等动物用品的来源与检测
功能实验室、化学及生物污染特殊区域的环境控制状况
清洁剂、消毒剂、杀虫剂使用情况
实验动物的来源、质量合格证明和检疫情况
六、仪器、仪表、量具、衡器等
七、机构主要仪器设备
八、SOP(制订、修改及废弃)
九、计算机系统运行和管理情况
十、YAO物安全性评价研究实施情况
GLP实验流程:
制定实验方案。
由项目负责人根据YAO物特点、YAO物安全性实验指导原则和GLP实施规定制定
将实验方案递呈质量保证部门审查,同时检查以往的SOP是否适应于该项实验。
实验进行中各业务部门实验工作记录、执行实验方案和SOP实施情况。
GLP实验流程(续)
总结报告:收集实验记录、对资料和数据进行处理,计算统计学及生物学意义;归纳YAO物毒性的主要靶器官,及临床上可能出现的毒性反应。
资料存档:将实验方案、各个环节的原始记录、各种标本及总结报告保存于档案部门。
软件要求:
1.建立完善的组织管理体系
2.高素质的工作人员队伍
3.对各项工作必须采用的标准操作规程(SOP)
4.对实验的全过程要求详细的进行记录
5.对数据管理与统计分析要求
6.组织严密的实验全过程的监督、检查和审查
硬件要求
:
1.对实验环境中各种设施的构造、宽广度、配置等有严格要求
2.对实验数据的收集、测定或分析等使用的仪器设备以及控制设备内环境有严格要求
GLP的基本内容和要求:
(一). 研究课题的组织与管理
(二). 对实验设施和仪器设备的规定与要求
(三). 标准操作程序(SOP)的制定和执行
(四). 实验设计方案(Protocol)及其主要内容
(五). 研究资料的收集、整理和报告
(一)研究课题的组织和管理 :
1. 业务/行政主管及其职责
任命,指定和更换课题组长
确保质量保证单位履行职能
提供人力,物力,财力等方面的保证
确认各种错误或过失得以纠正
对研究人员和质保单位之间的争议作出裁决
2. 课题组长及其职责
:
设计签署实验方案及其它正式文件并且注明日期。
-保证所有实验资料,包括意料之外的试验反应,都被准确地记录并核实。
-当实验过程中某些可能影响研究质量和结果一致性性的未预料到的情况发生时,及时采取措施(行动)补救,并且立即如实记录。
-确保GLP的各项规定都遵守执行。保证所有的原始资料、文件、实验设计、标本及正式实验报告按档案材料保存和管理。
签署正式研究报告,按照国家法规规定签署遵守声明(Compliance Statement)。
3. 质量保证单位及其职责 :
保存实验机构的Master Schedule,Protocols和SOPs(各1份)。
研究进行期间对研究课题多次监察,以确保研究的一致性。记录和签署每次监察的结
重要问题和发现立即口头知会课题长或提交书面报告,陈述所发现的问题以及采取的相应措施
审定无未经批准或记录的偏离Protocol和SOP的情况发生或出现
审校正式实验报告,确保报告中对研究方法SOP的描述准确无误,实验结果确实是原始资料整理分析的结果
拟写和签署附在正式报告的质量保证声明
(二)实验设施和仪器设备的规定要求
1.实验设施
:
实验机构具备足够的实验动物房间或者其他试验系统区域,以使
(1)不同种属的动物或试验系统分别饲育
(2)不同的课题分离饲育
(3)Quarantine动物
(4)建立常规或特别的动物饲育系统。
实验机构应有足够的动物房间或其他试验系统区域,以分别隔离那些用已知有生物危害的试验系统、受试物、对照物或参照物,wei xin:wlayar包括易挥发物质、气溶胶、放射活性材料以及感染性(传染性)微生物。
隔离的房间或区域应有诊断、处理和控制试验系统/动物疾病的装置。应有效地隔离有疾病、疑有疾病、或疾病携带的试验系统/动物的房间或区域。
1.实验设施(续)
实验设施应有和排放污染的水、土壤或其它材料的能力。如存养动物,则应有收集和处理所有动物废物和排泄物的能力,或者具有安全消毒的装置。
实验设施应按实验设计方案要求,具备调控环境条件如温度、温度、光线等能力。
2. 仪器设备
:
关键仪器设备 (critical equipment)
常规仪器设备
安全设施与设备
仪器设备的认证。计量审计及日常维修保养
三)SOP的制定和执行
1 . 种类和制定过程
标准操作程序(Standard Operating Procedure, SOP) 凡是实验设计方案中没有详细说明的常规方法和操作程序,都应该制SOP。GLP明确规定,至少需要就以下具体方法和操作步骤制定标准操作程序:
1. 试验系统/动物的准备
2. 试验系统/动物的饲养管理
3.受试物、对照物和参照物的接收、鉴别、贮放、配制,以及采样方法;
4. 对试验系统的观察;
5.(试)验和测试(定)方法
6. 垂(频)死中死亡试验系统/动物处理;
7. 试验系统的尸检或者尸解;
8. 标本的收集采取与标记;
9. 组织病理学检查;
10. 文件资料的处理、存放和检索;
11. 仪器设备的校正(准)和保养;
12. 试验系统的转移(运)、取(替)代和鉴别方法。
2. SOP的格式和内容 :
-每一种标准操作程序都必须有各自的编号(码),并且署明部门、版本和副本数目;
-标准操作程序的名称(题目)和生效日期;
-审批者和共(会)签者包括质量保证部门负责人的签字及签署日期;
-该标准操作程序的目的,涵盖范围,以及文中所用术语的定义;
-材料,操作程序和过程;
-转换和计算方法;
-报告要求,并将文中“材料”部分和报告的格式(常用表格式)作为标准操作程序之附件;
-分发和保存的规定与要求;
-负责此标准操作程序的有效性,并且监督其遵守执行的人员;
-该标准操作程序的复审和修订计划;
-意外事件的补救对策略;
-保密要求;
-参考文献和附件,包括各种表格。
(四)实验设计方案及其主要内容:
实验设计方案(Protocol)的主要内容应该包括:
-课题名称和研究目的;
-受试物、对照物和参照物的名称、化学文摘号(CAS),或者其它代码;
-实验委托者(保证者)和实(试)验实施机构的名称和详细地址:
-实验起始和终止完成日期;
-实验动物/试验系统的选择及其理由;
-尽可能地写明试验系统的种属(系)、年龄、性别,以及试验系统的来源;
-试验系统的鉴别方法和鉴别程序;
-描述具体实验设计,包括控制偏倚方法;
-说明实验期间所用饮料,溶剂、以及用以助溶或混 悬的其它材料 。 对于饮料中可能出现的污染物,要详细说明这些污染物的特性,可能影实验结果的含量(浓度),以及实验过程中可以容许的含量(浓度);
-染毒途径及其选择理由;
-染毒剂量/浓度单位。受试物、对照物和参照物的给予方式和次数;
-试验、分析和测定(量)的方法和次数;
-需要保存的记录和资料;
-委托者(保证者)和课题组长的准备签署日期;
-对研究中所用的统计处理方法加以说明。
五)研究资料的收集、整理和报告
文字记录(包括表格)
1 原始记录应该用不裉色笔(最好是用黑色)及时记录,字迹应清晰易认。
2 写明观察日期和记录日期(最好为同一天),实验观察者须在每页上签字。
3 不得用各种涂改液。
4 不准涂改重描(Re--Writes),应重写。
5 允许改错,但只能用单线斜线在需改正处,并且签名、署日期,说明改正的原因或理由(常用代码示之)。
6 必须用装订成册的实验记录本。
7 原则上不得有空白页。所有空页(处)要斜划注明“空白”,或者在最后一页加以特别说明
8. 如某部分原如资料与多项课题有关,可以复印(制)副本。 但每份副本均须注明“原样副本”(一般盖章),由复印(制)人签名和署日期并且说明原件在何处存放。
自动收集的原始资料
1 自动记录的各种资料,应逐个记记录日期、时间和仪器操作人。
2 如需改动,同样不准原记录上“涂改”(Obscure)。要注明改动理由、改动人并署明改动日期。
(六)正式实验报告的基本内容和要求:
正式实验报告(Final Report)。原则上应有以下项目和内容:
1、实验实施机构的名称和地址:实(试)验的起如日期、完成日期或者终(停)止日期。
2、设计方案中确定的研究目的和程序以及研究过程中设计方案的任何修改和变更。
3、用于分析实验数据资料的统计方法(必须与设计方案的相应部分一致)。
4、受试物、对照物和参照物的名称,化学文摘号或其它编码。这些物质的强度、纯度、组成及其它理化特性,以及它们在实验条件下的稳定性和溶解度。
5、描述说明实(试)验方法、、试验系统和染毒剂量。对于试验系统的描述应包括动物数目、性别、体重范围、种属(系)、年龄和来源,以及标记和鉴别动物的方法等;染毒剂量应同时说明剂量、给毒次数和间隔时间,染毒途径和持续时间等。
6、描述和说明所有可能影响数据资料的质量和一致性的环境条件因素。
7、课题组长、业务主管和所有参加课题的专业人员的姓名。
8、数据资料的转换、计算及统计处理。资料的分析与总结,以及由资料分析而得出的结论 。
9、课题组所有专业人员,包括委托或保证单位的代表、在资料或标本收集之后参加分析和评价的专业人员的姓名,签字和签署日期。
10、标本、原始资料和正式报告和存放地点。
11、质量保证单位的声明(Statement)。
(七)研究资料的保管、存贮和检索
1. 指定档案保管人,并且在组织机构图上反映出来。
2. 确定档案材料的设施、区域、或者机构。
3. 明确有权进出档案材料保管区域的人员,限制其他人进出该区域。
4. 建立来该登记制度,借阅档案材料要严格登记。
5. 建立便于检索的索引系统。
6. 分类管理各类档案材料,严格执行保存其限的规定。
7. 保管区域的环境条件要适宜于档案材料的长期保存,应有防/火设备和防虫蛀措施,以及紧急情况下的转移疏散方案。
EPA (FIFRA)-规定从以下三条中选择最长时间为保存期限:
1、用于向报审产品研究或上市的研究档案,在所有的相关研究全部结束之前,都应妥善保管;
2、报审自向正式递交报审之日起,至少保存五年;
3、如该项研究未用于报审,则从研究结束或终止之日起至少存二年。
EPA(TSCA)
-1、除湿(鲜)标本外,自最后试验规则有效起,至少保存10年;
2、商讨方案“(Negotiated Test Agreement)”自该方法案公布之日起,至少保存10年;
3.其它研究档案材料,从向政府管理部门递交正式研究结果之日算,至少保存5年。
FDA(非临床研究)-规定从以下三条中择最短时间为规范保存期限:
1、用来支持或补充已经批准研究和上市产品的档案材料,自批准之日起,最少保存2年;
2、研究的档案材料,自递交正式申请之日起,最少保存5年;
3、未能作为报审材料的档案,在实(试)验结束或终止后,再保存5年以上
第3节 实验设计的随机、对照、重复原则
实验设计的目的
1.有目的、有计划、有步骤地进行实验。
2.最大限度地减少实验误差。
3.节省时间、样本、经费开支,减少人力物力财力,获得较高的效率
对照的原则:
-意义:消除系统误差,鉴别处理和非处理因素的差异
-类型:空白对照、阳性对照、阴性对照、自身对照
随机的原则:
- 对照组和处理组除处理因素不同外,其余应全部相同。
- 动物实验随机分组;观察切片也应随机选择视野

重复的原则:
- 随机原则可排除大部分非处理因素的影响,但不能全部消除;因此,有必要重复,以排除偶然因素的影响。
- 表现为一定的试验样本数(人、动物、细胞株)
深圳市肯达信企业管理顾问有限公司-总部
- 公司类型私营独资企业
- 经营模式商业服务-私营独资企业
- 联系人王小姐
- 联系手机15626565226
- 联系固话0755-89335156-606
- 公司地址深圳市龙岗区平湖华南城五号2C071
-
 潍坊GMP认证办理山东GMP认证检测机构淄博化妆品药品医疗器械GMP认证 ¥8000.00
潍坊GMP认证办理山东GMP认证检测机构淄博化妆品药品医疗器械GMP认证 ¥8000.00 -
 ISO 22716体系认证化妆品良好生产规范山东GMP认证,潍坊化妆品药品gmp认证 ¥8000.00
ISO 22716体系认证化妆品良好生产规范山东GMP认证,潍坊化妆品药品gmp认证 ¥8000.00 -
 北京办理医美诊所需要哪些条件? ¥200000.00
北京办理医美诊所需要哪些条件? ¥200000.00 -
 洗脸巾生产许可证办理纸巾湿巾生产卫生许可证条件 ¥8000.00
洗脸巾生产许可证办理纸巾湿巾生产卫生许可证条件 ¥8000.00 -
 药用低密度聚乙烯膜、袋 YBB00072005-2015药品包装材料检测中心 ¥1000.00
药用低密度聚乙烯膜、袋 YBB00072005-2015药品包装材料检测中心 ¥1000.00 -
聚酯/铝/聚乙烯药用复合膜、袋 YBB00172002-2015药品包装材料检测中心 ¥1000.00
-
包装材料溶剂残留量测定法YBB00312004-2015 中国包装科研测试中心 ¥1000.00
-
加热伸缩率测定法 YBB00292004-2015中国包装科研测试中心 ¥1000.00
-
剥离强度测定法 YBB00102003-2015药品包装材料检测中心 ¥1000.00
-
软性屏障膜抗揉搓性 YY/T 0681.12-2014中国包装科研测试中心 ¥1000.00
-
药用铝塑封口垫片通则 YBB00212004-2015中国包装科研测试中心 ¥1000.00
-
输液瓶用铝塑组合盖 YBB00402003-2015 中国包装科研测试中心 ¥1000.00
-
软性屏障膜和复合膜抗慢速穿刺性 YY/T 0681.13-2014中国包装科研测试中心 ¥1000.00
-
透气包装的密封泄漏 YY/T 0681.4-2021 药品包装材料检测中心 ¥1000.00
-
聚酯/铝/聚乙烯药用复合膜、袋 YBB00172002-2015中国包装科研测试中心 ¥1000.00



